前言
2022年8月,阿拉巴马大学伯明翰分校Lewis Shi团队在Nature Communications发表“Selective suppression of melanoma lacking IFN-γ pathway by JAK inhibition depends on T cells and host TNF signaling”,该研究利用IFNγR1敲除的黑色素瘤细胞,构建免疫检查点阻断剂(immune checkpoint blocker, ICB)耐药小鼠模型,发现ICB耐药的潜在靶点JAK1/2,其抑制剂以T细胞和肿瘤坏死因子依赖的方式选择性抑制IFN-γ信号缺失的黑色素瘤。
背景介绍
ICB是恶性肿瘤治疗领域的重大突破,尤其是在黑色素瘤中。肿瘤细胞会通过控制免疫检查点蛋白,如CTLA-4、PD-1、PD-L1等,来逃避免疫监视。ICB则通过阻断免疫检查点,在肿瘤浸润T细胞(tumor-infiltrating T cell, TIL)中增强免疫效应功能,并且降低免疫抑制的FoxP3+调节性T细胞(Treg)的丰度,从而实现对肿瘤的抑制。
尽管如此,仍有许多患者对ICB治疗耐受,其中一个主要原因是肿瘤细胞IFN-γ信号缺失,目前尚缺乏针对ICB耐药的治疗策略。肿瘤内IFN-γ信号在ICB抗肿瘤功能中是必需的,但在作者的前期研究中,对黑色素瘤敲低IFNγR1(IFNγR1KD)却并没有明显改变TIL中CD8+/Treg比例,这可能由于细胞中仍有残余的IFN-γ信号。作者考虑利用CRISPR技术对IFNγR1进行敲除,进而探究克服ICB耐药的潜在靶点。
研究思路
01 构建IFNγR1敲除的黑色素瘤细胞系,以及移植瘤小鼠模型,验证模型能够有效模拟IFN-γ信号缺失导致的ICB耐药。
02 从肿瘤免疫微环境分析ICB耐药,检测IFN-γ信号缺失对TIL的影响,发现IFN-γ信号缺失使肿瘤内T细胞特征减少。
03 利用激酶组分析寻找可能的ICB耐药靶点,发现IFN-γ信号缺失的黑色素瘤细胞中JAK1/2激酶通路的激活。
04 分别从胞外和胞内探究JAK1/2通路激活的因素,找到PI3K-Akt和mTOR信号通路,通过不同抑制剂处理等证明mTOR在上游调控JAK1/2通路。
05 体内验证JAK1/2抑制剂可以通过T细胞和TNF因子依赖的方式特异性阻碍IFN-γ信号缺失的黑色素瘤生长。
研究内容
首先,作者利用CRISPR-Cas9技术在B16细胞中敲除IFNγR1(IFNγR1KO),并接种至BL6小鼠中构建黑色素瘤小鼠模型。经验证IFNγR1KO细胞对IFN-γ的刺激没有响应,表现为JAK2磷酸化、Irf1、PD-L1、MHC-I/II等在IFN-γ刺激下均没有上调,此外IFN-γ也不能诱导IFNγR1KO细胞凋亡或抑制其细胞增殖。IFNγR1的敲除本身对B16细胞体内成瘤能力没有影响,但IFNγR1KO黑色素瘤对CTLA-4抗体的处理耐受。与体外数据一致,体内IFNγR1KO黑色素瘤经CTLA-4抗体治疗后,PD-L1和MHC-II等表达也没有上调。
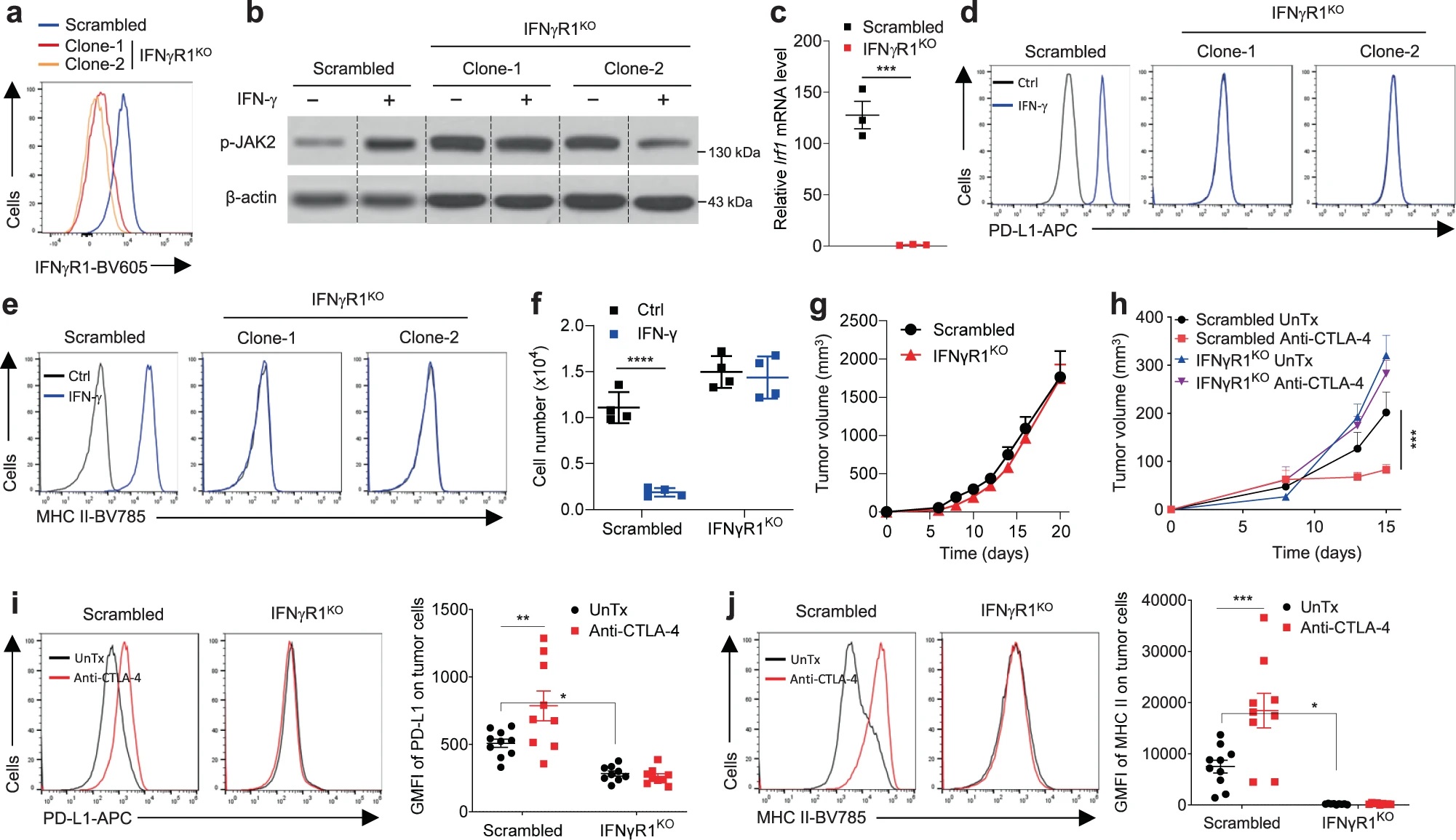
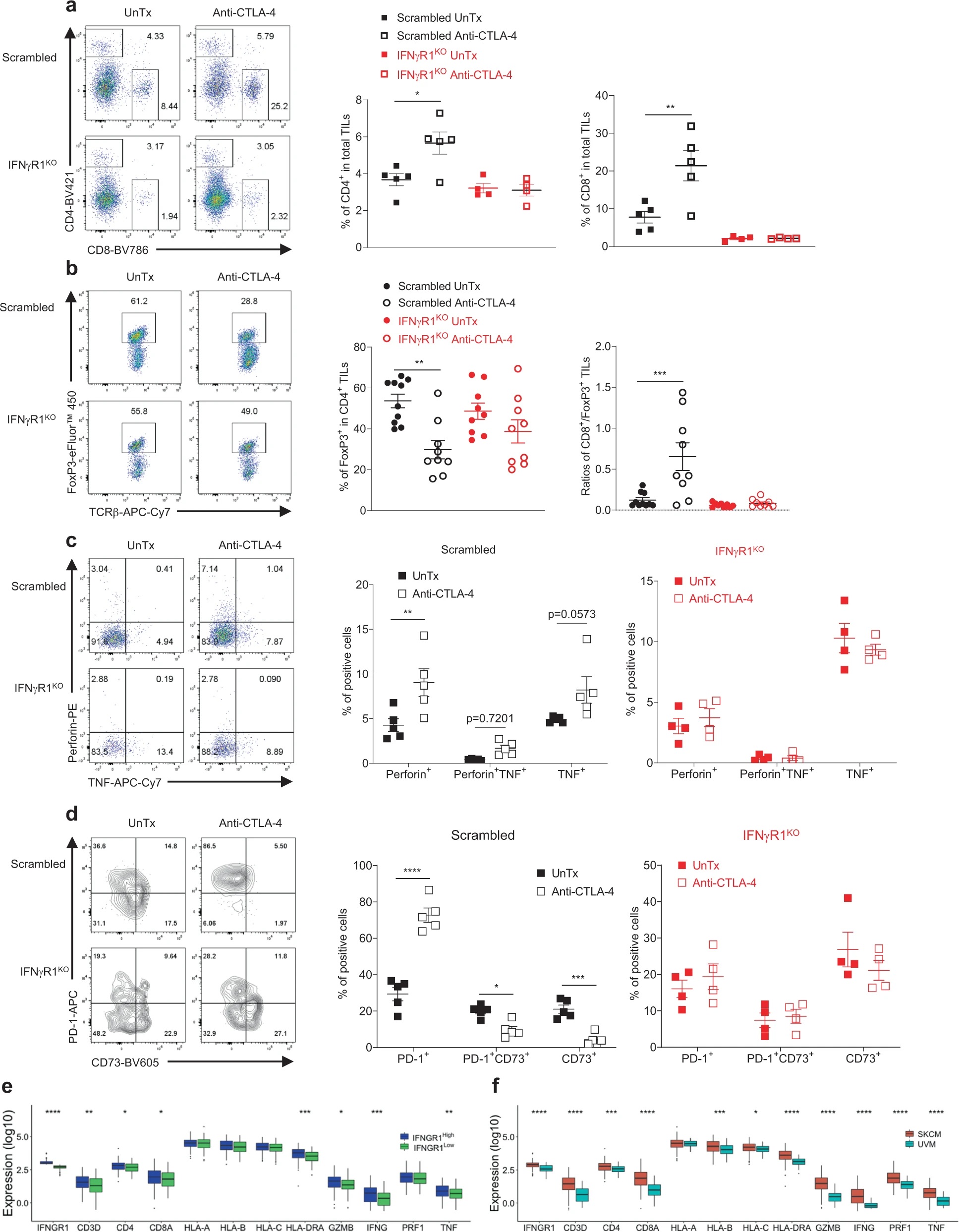
接下来作者分析IFNγR1KO黑色素瘤中的TIL表型。流式发现CD8+ T细胞的基数减少,并且CTLA-4抗体治疗不会像对照黑色素瘤那样,增加T细胞浸润、减少瘤内Treg细胞比例、或促进效应细胞因子分泌等等。对TCGA数据库中黑色素瘤进行分析,按照IFNGR1表达量分为两组,发现IFNGR1低表达的黑色素瘤中T细胞特征基因表达降低,包括T细胞表面标志物、效应分子和MHC分子等,同时IFNGR1低表达的黑色素瘤患者生存率也显著降低。相比于皮肤黑色素瘤(SKCM),葡萄膜黑色素瘤(UVM)对免疫检验点抑制剂(ICB)更加耐受,数据库表明UVM中IFNGR1表达,以及T细胞特征基因表达更低。
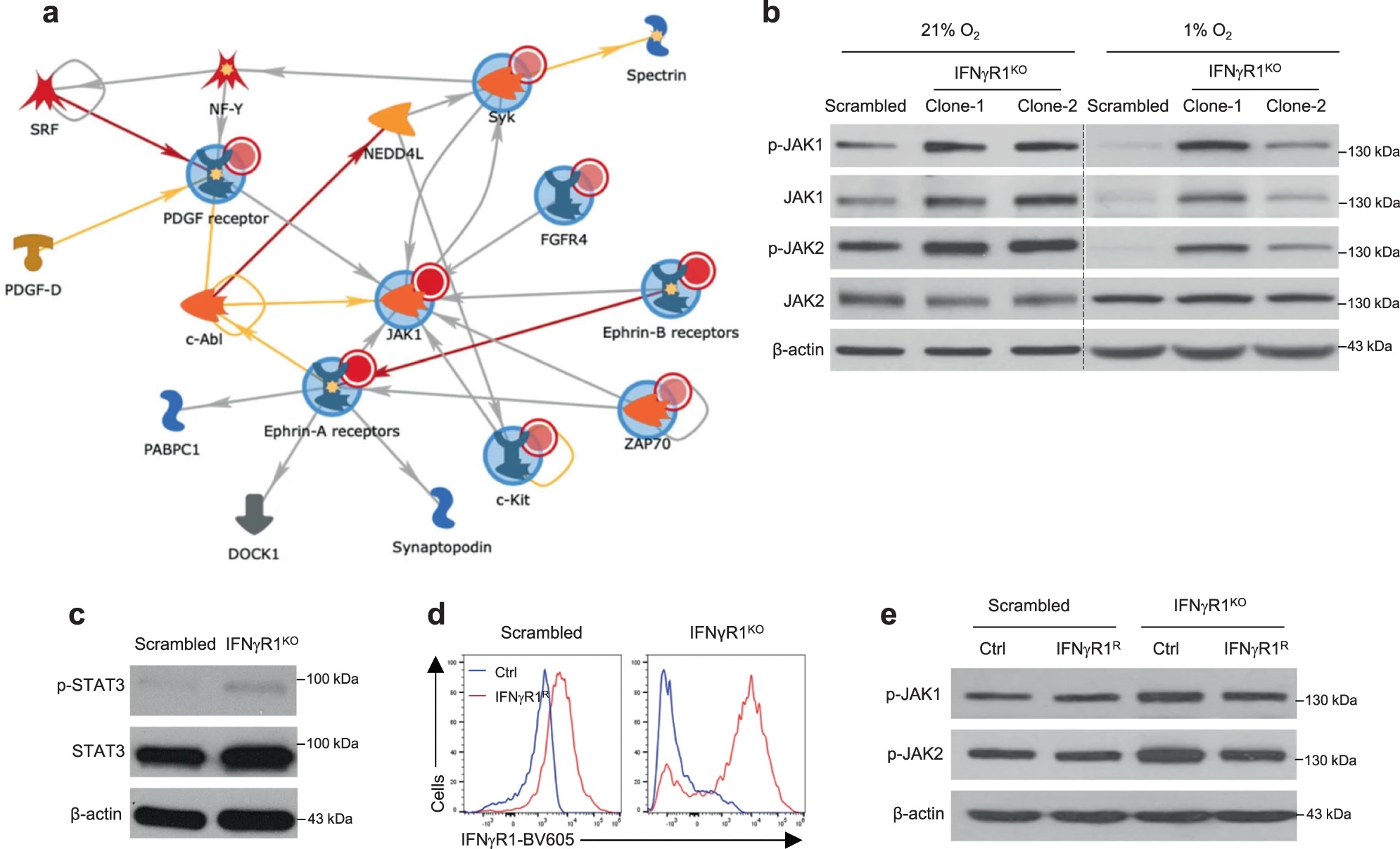
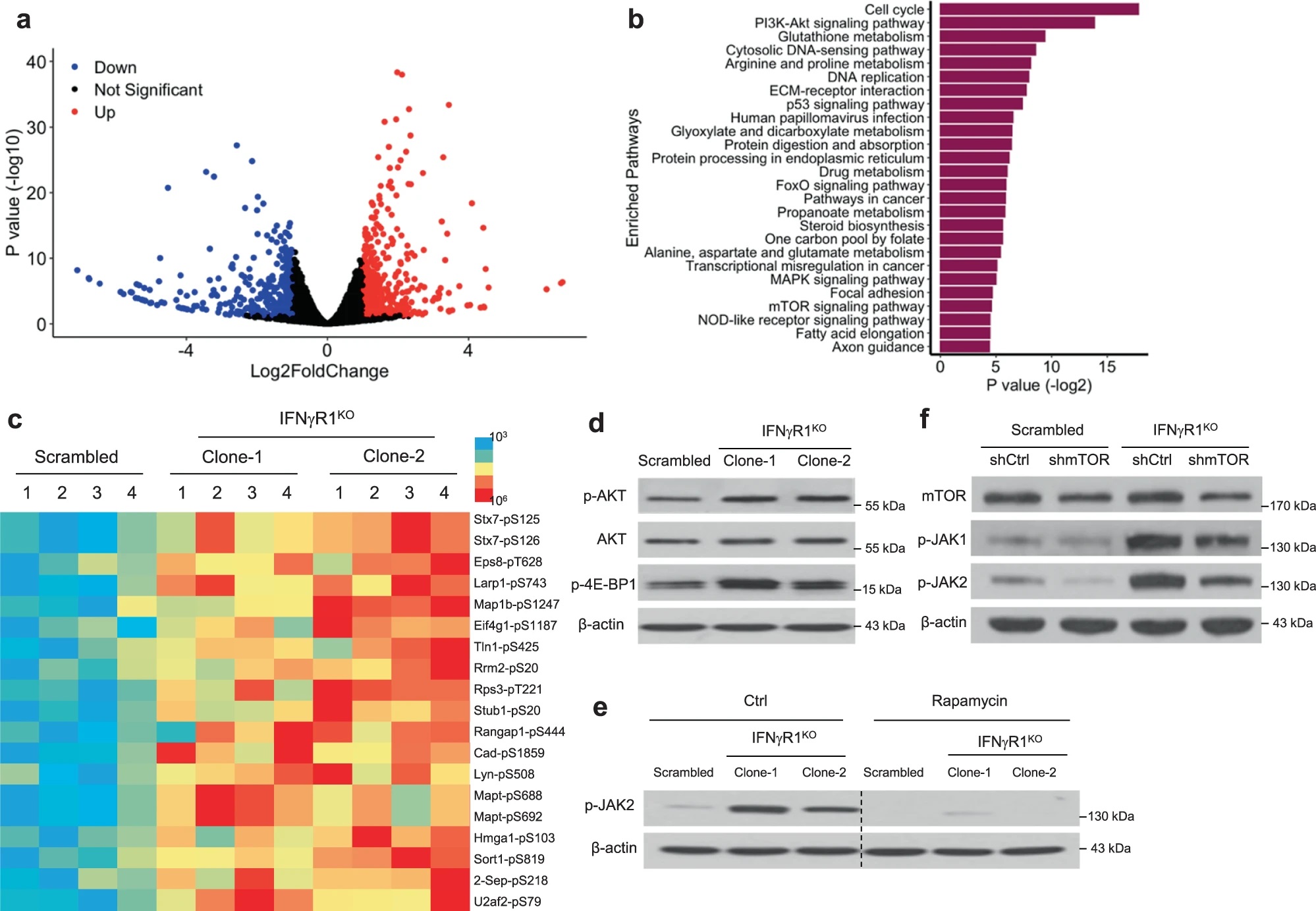
考虑到酪氨酸激酶在IFN-γ信号中的重要作用,作者对IFNγR1KO细胞进行了激酶组活性分析,试图在其中寻找克服ICB耐药的靶点。这一分析发现激活的激酶网络汇集到JAK1/2,WB验证IFNγR1KO细胞JAK1/2及其下游STAT3的磷酸化水平升高,而在IFNγR1KO细胞重新表达Ifngr1后,JAK1/2磷酸化水平回调,说明黑色素瘤中IFN-γ信号的缺失与JAK1/2异常激活存在联系。
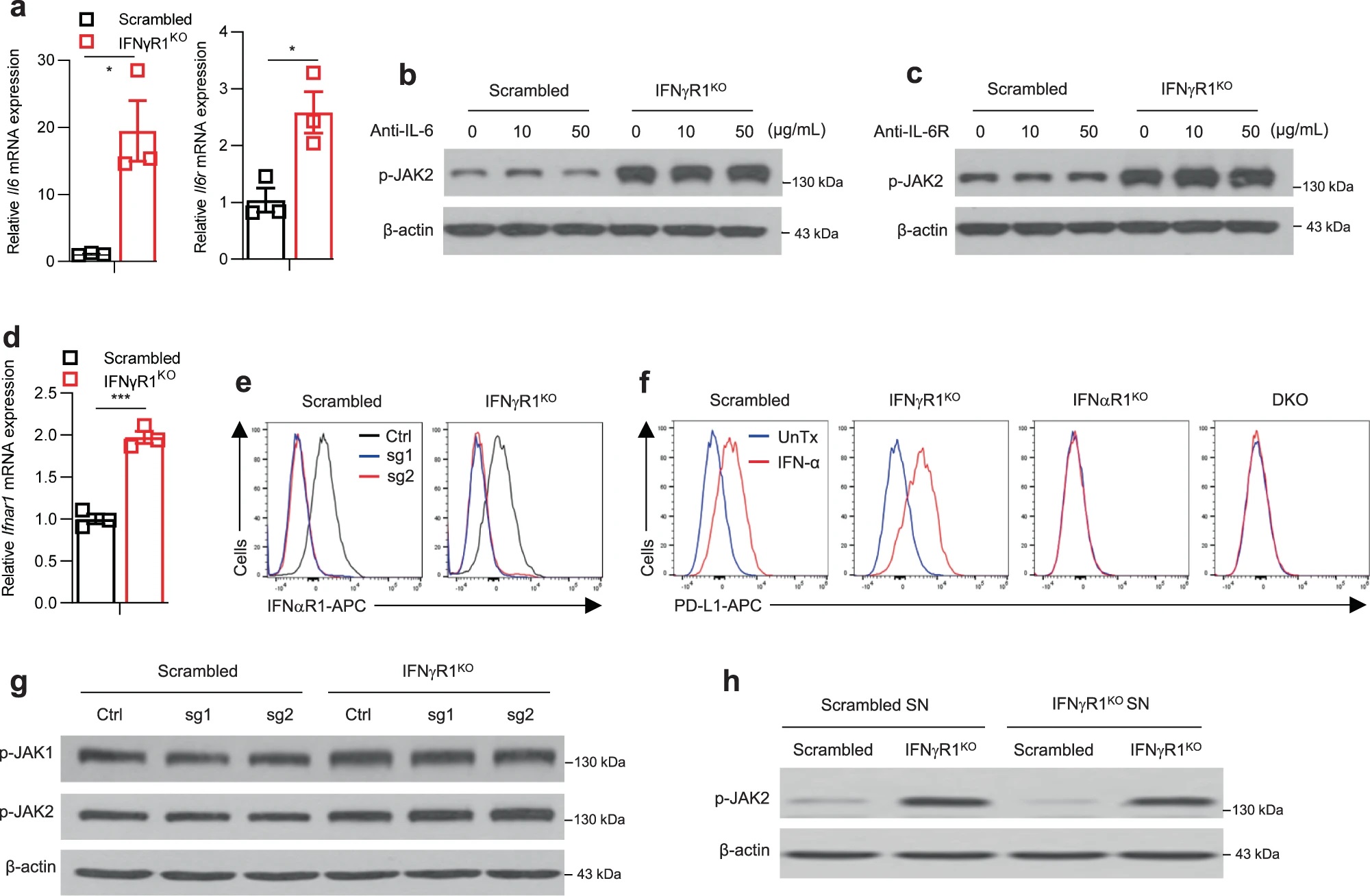
JAK-STAT通路可能由胞外信号如IFN-γ、IFN-α/β、IL-6等激活。Il6、Il6r以及IFNαR1(IFN-α/β受体)在IFNγR1KO细胞中表达升高,但IL-6和IL-6R抗体处理,或敲除Ifnar1均不能使IFNγR1KO细胞的JAK1/2磷酸化水平降低,说明不是这些因素使JAK1/2激活。进一步分析其他胞外因素,作者用IFNγR1KO细胞的培养基上清处理对照黑色素瘤细胞,发现并不能诱导JAK2磷酸化水平上升,因此IFNγR1KO细胞JAK1/2的激活更可能是胞内事件的结果。
为了明确使JAK1/2的激活胞内因素,作者对IFNγR1KO和对照细胞进行了全转录组测序,利用差异表达基因进行通路富集分析,并且进行了磷酸化蛋白质组学研究。这些分析均发现PI3K-Akt和mTOR信号通路的显著差异,WB验证了IFNγR1KO细胞中AKT和4E-BP1(mTOR下游)磷酸化水平升高。作者进一步研究JAK1/2与mTOR通路的关系。mTOR抑制剂rapamycin (Rapa)处理IFNγR1KO细胞显著抑制JAK2磷酸化,而JAK1/2抑制剂ruxolitinib (Ruxo)处理后4E-BP1磷酸化水平不变,并且在IFNγR1KO细胞中敲低mTOR也能使JAK1/2磷酸化回调。因此,在IFN-γ信号缺失的黑色素瘤细胞中,mTOR通路信号增强,并调控下游JAK1/2的激活。
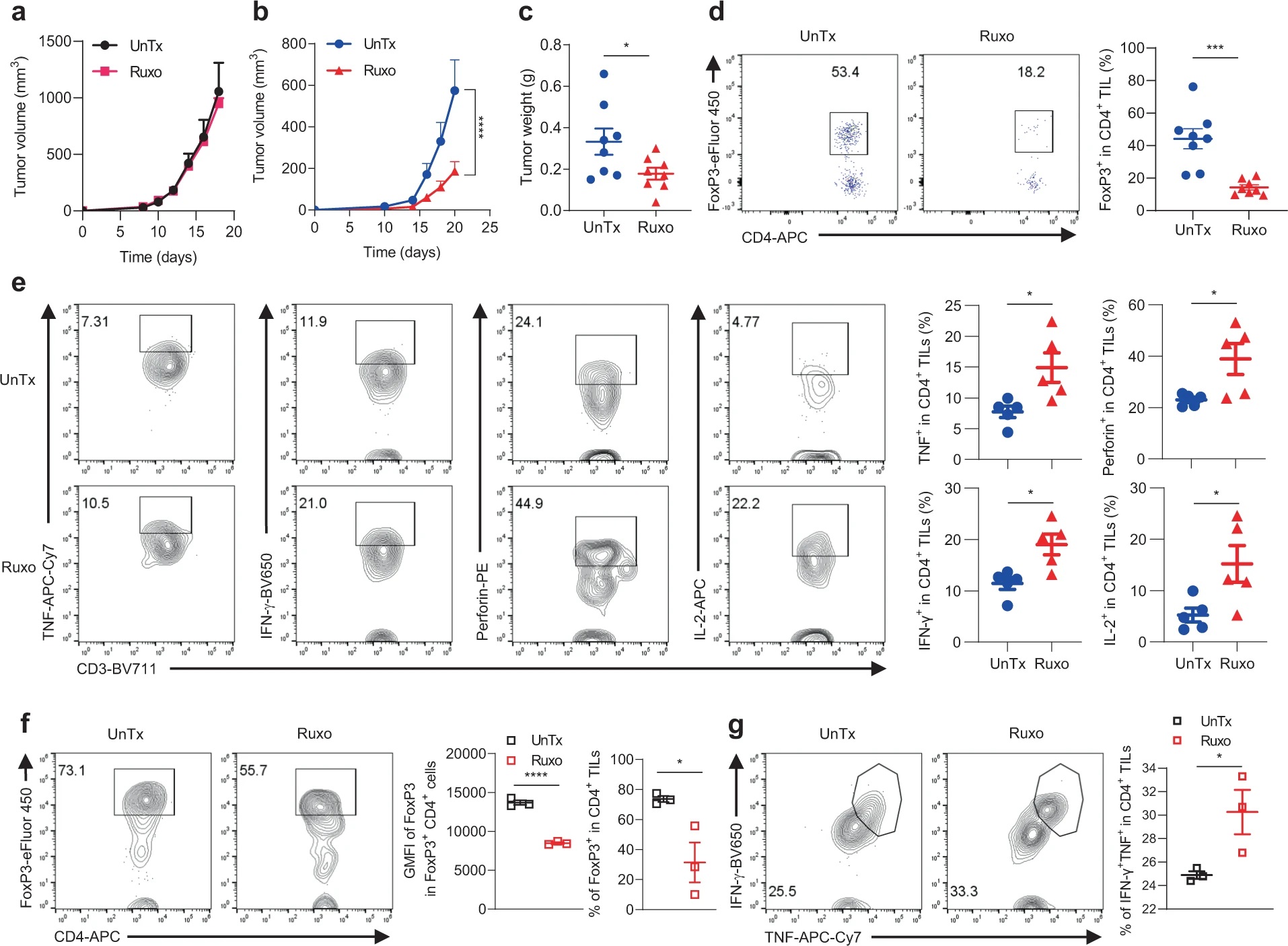
上述结果提示IFN-γ信号缺失导致的ICB耐药或许可以通过靶向mTOR-JAK1/2来解决,作者随后在B6小鼠中进行体内验证。在用Ruxo分别处理IFNγR1KO和对照黑色素瘤小鼠模型后,发现Ruxo仅特异性抑制IFNγR1KO黑色素瘤生长。尽管如此,体外实验表明这一抑制作用并非由Ruxo对IFNγR1KO细胞的直接杀伤所致。对体内黑色素瘤的分析显示,Ruxo能够减少CD4+ TIL中Treg细胞比例,增加TNF、IFN-γ、穿孔素、IL-2的分泌。而从未经药物处理的黑色素瘤中分离TIL进行体外培养,发现Ruxo处理降低FoxP3表达,增强TIL的效应功能。以上结果表明Ruxo依赖于TIL发挥对IFN-γ信号缺失的黑色素瘤的抑制作用,而非直接杀伤肿瘤细胞。
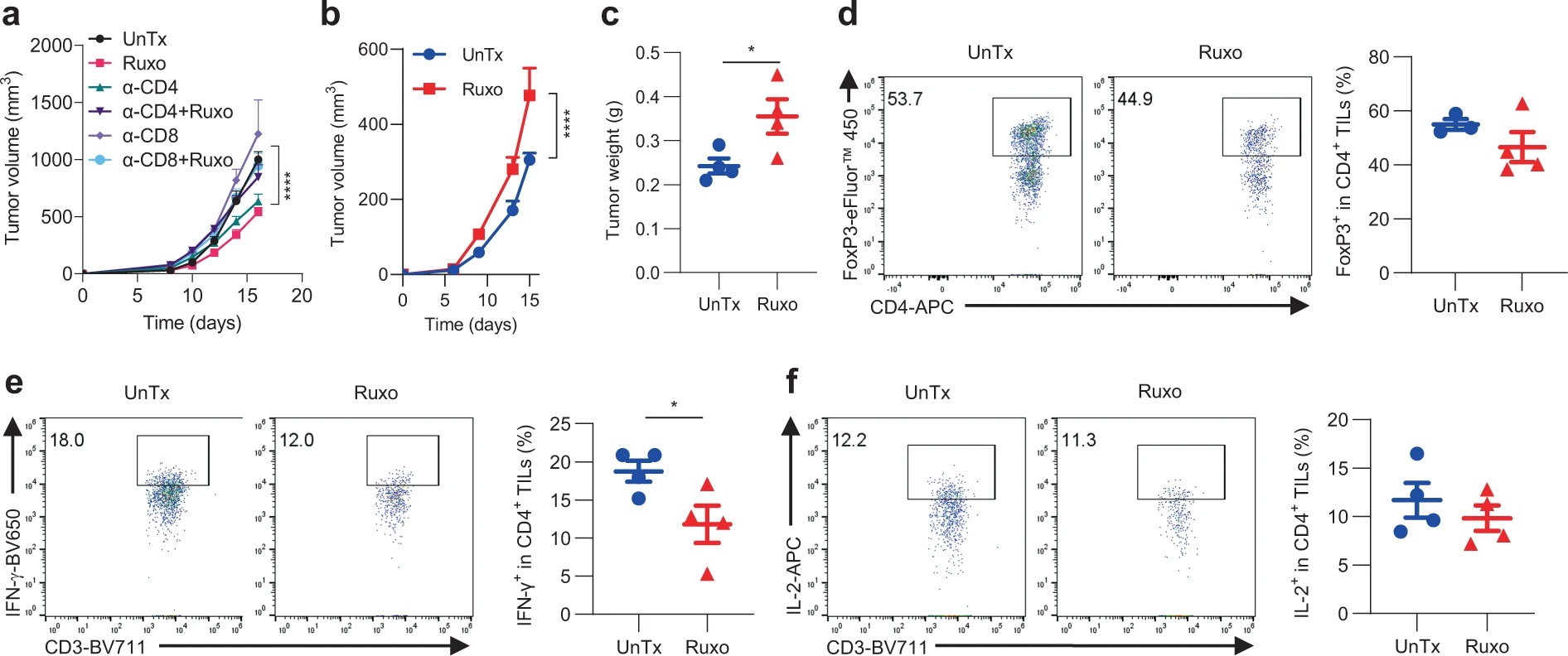
为了评估T细胞在Ruxo治疗中的作用,作者对接受Ruxo治疗的IFNγR1KO黑色素瘤小鼠进行CD4或CD8中和抗体处理,发现任一种T细胞的缺失都使Ruxo的治疗无效。在对Ruxo抑癌效应机制的探究中,作者关注到TNF。IFNγR1KO黑色素瘤细胞接种至TNF敲除(TNF-/-)小鼠后,Ruxo不能将其抑制。对TIL的分析也发现,TNF-/-小鼠中Ruxo处理不能使Treg细胞比例下降,或使IFN-γ、IL-2等因子分泌增加。因此,Ruxo对IFNγR1KO黑色素瘤的特异性抑制依赖于T细胞和TNF因子。
综上所述,文章利用IFNγR1敲除的黑色素瘤小鼠模型,发现IFN-γ信号缺失的黑色素瘤免疫微环境更不利于抑制肿瘤;结合激酶组、转录组、磷酸化蛋白质组等多组学分析,发现此类黑色素瘤中mTOR通路信号增强,从而激活下游JAK1/2激酶;JAK或许可以作为IFN-γ信号缺失所致ICB耐药的靶点,对临床有一定的指导意义。
和元生物凭借多年技术积累,基于基础的病毒服务和公司自有的细胞、动物平台,我们可以为您提供慢病毒包装、细胞株构建、流式分析及分选、小鼠移植瘤模型构建和包括活体成像在内的各种动物操作服务,为您提供文中涉及的多种研究支持。