
质粒提取经常因操作不当引起提取的质粒不纯,或遇到各种问题,
本文归纳了几个质粒提取过程中常见的问题及解决方法供大家参考。
Q1:涂布棒在酒精蘸一下,然后烧一下,能不能保证把所用的菌烧死?
小科回答:涂布棒可以在酒精中保藏,但是酒精不能即时杀菌。蘸了酒精后再烧一小会,烧的是酒精而不是涂布棒。建议涂布棒干烧较长时间后,冷却后再涂。同时作多个转化时,应用几个涂布棒免得交叉污染。
Q2:原先测序鉴定没有问题的细菌,37℃摇菌后发现质粒大小或序列出现异常?
小科回答:这种情况出现的几率较小,常出现在较大质粒或比较特殊的序列中。解决办法:
(1)降低培养温度,在20~25℃下培养,或室温培养可明显减少发生概率;
(2)使用一些特殊菌株,如Sure菌株,它缺失了一些重组酶,如rec类等,使得质粒复制更加稳定;
(3)质粒抽提有一个酶切不完全的原因就是溶液Ⅱ中的NaOH浓度过高造成的。
Q3:未提出质粒或质粒得率较低,如何解决?
小科回答:
(1)大肠杆菌老化:涂布平板培养后,重新挑选新菌落进行液体培养;
(2)质粒拷贝数低:由于使用低拷贝数载体引起的质粒dna提取量低,可更换具有相同功能的高拷贝数载体;
(3)菌体中无质粒:有些质粒本身不能在某些菌种中稳定存在,经多次转接后有可能造成质粒丢失。例如,柯斯质粒在大肠杆菌中长期保存不稳定,因此不要频繁转接,每次接种时应接种单菌落。另外,检查筛选用抗生素使用浓度是否正确;
(4)碱裂解不充分:使用过多菌体培养液,会导致菌体裂解不充分,可减少菌体用量或增加溶液的用量。对低拷贝数质粒,提取时可加大菌体用量并加倍使用溶液,可以有助于增加质粒提取量和提高质粒质量;
(5)溶液使用不当:溶液2和3在温度较低时可能出现浑浊,应置于37℃保温片刻直至溶解为清亮的溶液,才能使用;

(6)吸附柱过载:不同产品中吸附柱吸附能力不同,如果需要提取的质粒量很大,请分多次提取。若用富集培养基,例如TB或2×YT,菌液体积必须减少;若质粒是非常高的拷贝数或宿主菌具有很高的生长率,则需减少LB培养液体积;
(7)质粒未全部溶解(尤其质粒较大时) :洗脱溶解质粒时,可适当加温或延长溶解时间;
(8)乙醇残留:漂洗液洗涤后应离心尽量去除残留液体,再加入洗脱缓冲液;
(9)洗脱液加入位置不正确:洗脱液应加在硅胶膜中心部位以确保洗脱液会完全覆盖硅胶膜的表面达到最大洗脱效率;
(10)洗脱液不合适:DNA只在低盐溶液中才能被洗脱,如洗脱缓冲液EB(10mM Tris-HCl, 1mM EDTA,pH8.5)或水。洗脱效率还取决于pH值,最大洗脱效率在pH7.0-8.5间。当用水洗脱时确保其pH值在此范围内,如果pH过低可能导致 洗脱量低。洗脱时将灭菌蒸馏水或洗脱缓冲液加热至60℃后使用,有利于提高洗脱效率;
(11)洗脱体积太小:洗脱体积对回收率有一定影响。随着洗脱体积的增大回收率增高,但产品浓度降低。为了得到较高的回收率可以增大洗脱体积;
(12)洗脱时间过短:洗脱时间对回收率也会有一定影响。洗脱时放置1min可达到较好的效果;
Q4、细菌离心加入溶液I蜗旋振荡后,发现菌体呈絮状不均匀或呈细砂状?
小科回答:推荐联科生物快速质粒DNA小量纯化试剂盒,创新的独特buffer N8,2分钟内获得无沉淀的上清液而且颜色变化指示蛋白沉淀是否完全。
造成这种现象,可能的原因及解决方法:
(1)很可能是细菌发生溶菌,可减少培养时间或者试试平板培养,质粒提取前用PBS将菌落洗下,相较来说固体培养基上细菌生长的要好一些;
(2)质粒抽提过程很大程度上是受细菌生长情况决定,刚活化的菌比-80℃保存菌种所培养出来的菌液状态好,保存久的菌株可能会造成质粒浓度低,质粒丢失等不明原因;
(3)判断生长的菌液是否正常,可以用肉眼观察,在光线明亮处摇荡新鲜培养液,如果发现菌液呈漂絮状,情况很好。如果发现呈泥水状,即看不到絮状,只是感觉很浑浊,则可能提不出好的质粒,或者没有质粒;
(4)菌液不宜生长太浓,摇床速度不宜过高,达到OD600 1.5就可。
联科生物快速质粒 DNA 小量纯化试剂盒
► 核酸纯化柱无须平衡液处理;
► 8分钟内即可完成质粒DNA的制备。
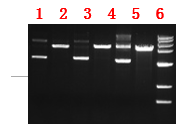
抽提不同质粒及其酶切,酶切条带干净,无其它非特异条带。
|
图注:
1:pET28a 2:pET28a的EcoR I单酶切
3:pET30a 4:pET30a的EcoR I单酶切
5:pGEX4T1 6:pGEX4T1的EcoR I单酶切 |
 |
| 目录号 | 规格 | 目录价格 |
| MKF01050401-050 | 50T | 150 |
| MKF01050401-250 | 250T | 650 |
以上文章来源生物帮。